News & Announcements
Drexel University and Thomas Jefferson University are longtime allies in the fight against cancer. That partnership was formalized in 2013 as the Sidney Kimmel Cancer Center (SKCC) Research Consortium, a relationship that was reinvigorated in 2021, in preparation for the center’s application to be named a National Cancer Institute-designated comprehensive cancer center. Read more. (Pulse Spring 2024)

Drs. Kenny Simansky and Noreen Robertson
Lifetime Achievement Awards
During the Sidney Kimmel Cancer Center retreat on Monday Feb 5, 2024, Drs. Kenny Simansky and Noreen Robertson were awarded the prestigious Lifetime Achievement Award!
Members of the College of Medicine community came together on Thursday, November 2 at the Pennsylvania Convention Center in Center City Philadelphia for Discovery Day 2023. The annual event highlights participants’ original research across many biomedical science and clinical research topics. Read more.
How can you best connect with peers and mentors? What are some good places to eat, study and relax — on or off campus? Current graduate students shared tips for Dragons who are just starting at the University this year. Read more.
Last term, Drexel University professors were recognized for their scholarly research and prolific academic and professional contributions. This update offers a snapshot of activity courtesy of the Office of the Provost. Read more.
We are pleased to announce that Asif M. Ilyas, MD ’01, MBA, FACS, has accepted the newly created position of associate dean of clinical research at Drexel University College of Medicine. Read more.
The Future of MRNA Therapeutics
On Wednesday, May 4, 2022, Franklin Institute Award laureates Katalin Karikó, PhD and Drew Weissman, MD, PhD presented a special lecture titled "The Future of MRNA Therapeutics." Drs Karikó and Weissman are recipients of the 2022 Benjamin Franklin Medal in Life Science. Katalin Karikó, PhD, is a senior vice president at BioNTech, and an adjunct professor of neurosurgery at the Perelman School of Medicine, University of Pennsylvania. Drew Weissman, MD, PhD, is the Roberts Family Professor of Vaccine Research at the Perelman School of Medicine, University of Pennsylvania. View presentation. (Drexel credentials required to access.)
Drexel Sustains Prestigious R1 Research Classification
We are proud to share the exciting news that Drexel University has again achieved national recognition in the Carnegie Classification of Institutions of Higher Education, retaining its designation as an “R1 Doctoral University: Very High Research Activity.” Drexel is one of 146 institutions out of approximately 3,900 to receive this prestigious classification, indicating the highest level of research activity. Notably, we are one of only 39 private universities to earn the distinction.
Members of the College of Medicine community came together this week for Discovery Day 2021, gathering at the Philadelphia Convention Center to discuss original research. The Thursday, October 21 event showcased a variety of biomedical science and clinical research topics. Read more.
Two Drexel researchers received prestigious Individual Biomedical Research Awards from The Hartwell Foundation to support their work aimed at benefitting the health of children of the United States. Each award includes $100,000 in research funding per year for three years. Read more.
For the second consecutive year, the Medical Student Summer Research Fellowship (MSSRF) Program will support 50 first-year medical students in full-time summer research. Prior to the COVID-19 pandemic, MSSRF had capacity to provide opportunities to 20 students each year. Read more.
After receiving approval by the Pennsylvania Department of Health to conduct research on medical cannabis grown by a clinical registrant, and then signing an eight-year, $15.5 million contract with the clinical registrant, the Chester-based Agronomed Biologics LLC, Drexel University has now opened a new Medical Cannabis Research Center (MCRC) to begin conducting evidenced-based research on effects medical cannabis has on patients with specific medical and behavioral maladies. Read more. (DrexelNow)
Fighting a global pandemic requires innovation and urgency, and Drexel’s researchers have heeded the call. Vaccine adjuvants, infection blockers and protein targeting to inactivate the virus are among the 17 COVID-19 projects currently supported by the $100,000 Rapid Response Research and Development Fund, created by University trustees. More than half the awarded projects involve College of Medicine faculty. (Pulse Fall 2020) Read more.
The pursuit for more information about the COVID-19 coronavirus has led a group of College of Medicine researchers to West Reading, Pa. and back again, with some of their vital work taking place before the sun is up. Read more.
COVID-19 Research Forum
May 14, 2020
Part 1: Clinical Framework, Translational Science and Population Science
12:00 - Welcome and Opening Remarks
Charles B. Cairns, MD, Walter H. and Leonore Annenberg Dean, DUCOM and Senior Vice President for Medical Affairs, Drexel University Professor of Medicine and Emergency Medicine
Introduction to the Research Forum Series
Kenny J. Simansky, PhD, Moderator Vice Dean for Research, Professor of Pharmacology and Physiology, Professor of Psychiatry
12:05 - COVID-19: Clinical presentation and epidemiology
Debra Powell, MD, MS, Chief, Division of Infectious Disease, Medical Director for Infection Prevention, Tower Health
12:15 - Translational Science Projects: Awards to DUCOM Principal Investigators from the Drexel University Rapid Response Research and Development Fund (Complete authorship information at drexel.edu/research/resources/response-to-covid-19-pandemic/fundingopportunities/rapid-response-research-development-funding-awardees/)
Adenosine Deaminase-1 Enhances Efficacy of a DNA Vaccine Encoding SARS-CoV-2 Spike Glycoprotein S1 Through Modulation of T-Follicular Helper Cells
Michele Kutzler, PhD, Assistant Dean of Faculty Development, Associate Professor of Medicine, Associate Professor of Microbiology and Immunology
Irreversible Inactivation of SARS-CoV-2 by Spike Protein Targeting
Irwin Chaiken, PhD Professor of Biochemistry and Molecular Biology
SARS-CoV-2 Infection and Sensing in Primary Human Macrophages
Sonia Navas-Martin, PhD Associate Professor of Microbiology and Immunology
Q&A for Translational Science Projects
12:40 - Population Science: Immune Response and Disease Severity in Hospitalized COVID Patients
Program funded by National institute of Allergy and Infectious Diseases
Immunophenotyping Assessment in a COVID-19 Cohort (IMPACC Study)
Elias Haddad, PhD Director of MD/PhD Program, Professor of Medicine
Charles B. Cairns, MD Walter H. and Leonore Annenberg Dean, DUCOM and Senior Vice President for Medical Affairs, Drexel University Professor of Medicine and Emergency Medicine
Q&A for COVID-19 Immunophenotyping Study
COVID-19 Research Forum
May 21, 2020
Part 2: Diagnostics, Vaccines, Therapeutics, and Technology
12:00 - Opening Remarks: Recap of Part 1 and Foundation for Today's Forum
Brian Wigdahl, PhD Professor and Chair, Microbiology and Immunology Director, Institute for Molecular Medicine and Infectious Disease
12:10 - Technologies and Therapeutic Behavioral Intervention: Awards to DUCOM Principal Investigators from the Drexel University Rapid Response Research and Development Fund (Complete authorship information at drexel.edu/research/resources/response-to-covid-19-pandemic/fundingopportunities/rapid-response-research-development-funding-awardees/)
Novel Mobile App and Information System for COVID-19 Situational Awareness and Organizational Management
Charles B. Cairns, MD, Walter H. and Leonore Annenberg Dean, DUCOM and Senior Vice President for Medical Affairs, Drexel University Professor of Medicine and Emergency Medicine
PPE Fabric Face Mask for Healthcare Providers and General Use1
Charles B. Cairns, MD, Walter H. and Leonore Annenberg Dean, DUCOM and Senior Vice President for Medical Affairs, Drexel University Professor of Medicine and Emergency Medicine
1 Genevieve Dion, Principal investigator Director, Center for Functional Fabrics, Professor of Design, Westphal College of Media Arts & Design
Implementation of an Online Peer Support Community to Assist Women with Substance Abuse Disorder During the COVID-19 Pandemic: A Pilot Study
David Bennett, PhD Professor of Psychiatry
Q&A for Technologies and Therapeutic Behavior Intervention
Moderator: Kenny J. Simansky, PhD, Vice Dean for Research, Professor of Pharmacology and Physiology, Professor of Psychiatry
12:30 - Round-Table: Detection, Prevention and Cure
Elias Haddad, PhD, Moderator Director of MD/PhD Program, Professor of Medicine
12:30 - Introductory Comments
Detection of Viral Nucleic Acid, Antigen, and Antibodies
Alan Evangelista, MD, PhD, Director, Microbiology and Virology, Pathology and Laboratory Medicine, St. Christopher's Hospital for Children
Pipeline for SARS-CoV-2 Vaccine
Michele Kutzler, PhD, Assistant Dean of Faculty Development, Associate Professor of Medicine, Associate Professor of Microbiology and Immunology
Detection of Viral Nucleic Acid, Antigen, and Antibodies
12:40 - Round-table
Garth D. Ehrlich, PhD, Executive Director, Centers for Advanced Microbial Processing, Genomic Sciences and Surgical Infections and Biofilms, Institute for Molecular Medicine and Infectious Disease Professor of Microbiology and Immunology, Professor of Otolaryngology-Head and Neck Surgery
Alan Evangelista, MD, PhD, Director, Microbiology and Virology, Pathology and Laboratory Medicine, St. Christopher's Hospital for Children
Mark G. Martens, MD, Senior Vice President - Academic Affairs, Chief Academic Officer and Designated Institutional Official, Tower Health Interim Vice Dean of Drexel University School of Medicine at Tower Health, Associate Dean for Graduate Medical Education, Professor of Obstetric and Gynecology, DUCOM
Brian Wigdahl, PhD, Professor and Chair, Microbiology and Immunology Director, Institute for Molecular Medicine and Infectious Disease
12:50 - Q&A for Round-table Including Introductory Comments
Project Awardees — COVID-19 Rapid Response Research & Development Fund
Drexel's Rapid Response Research & Development Fund was designated for urgent action, short-term projects focused on COVID-19 related health and health-related research and development.
The enthusiasm and the level of response to the Rapid Response Research & Development Fund was incredible. We received 40 submissions. With only a limited amount of funding available, selecting projects amongst this strong group of submissions was very challenging.
The awarded projects fall into five broad categories, they are:
Connected, Public and Mental Health
- Chronicling the Impact of the COVID-19 Pandemic on Physical and Mental Health and Telehealth Care Delivery: Perspectives from Providers and Older Adults
♦ RoseAnn DiMaria-Ghalili (CNHP), Kym Montgomery (CNHP), Gloria Gonzalez-Kruger (CNHP), Michael Weingarten (CoM)
- COVID-19: Slow Disaster
♦ Scott Knowles (CoAS)
- Creating a COVID-19 Urban Vulnerability Data Tool to Inform Mobilization and Coalition-Building
♦ Amy Carroll-Scott (DSPH), Félice Lê-Scherban (DSPH)
- Implementation of an Online Peer Support Community to Assist Women with Substance Abuse Disorder during the COVID-19 Pandemic: A Pilot Study
♦ David Bennett (CoM), Barbara Schindler (CoM), Chris Yang (CCI), Ou Stella Liang (CCI)
- Improving Spatial Surveillance for COVID-19 Cases While Accounting for Errors in Testing
♦ Neal Goldstein (DSPH), Igor Burstyn (DSPH)
- Novel Mobile App and Information System for COVID-19 Situational Awareness and Organizational Management
♦ Charles Cairns (CoM)
- RAPID Assessment of First Responder Mental Health
♦ Jennifer Taylor (DSPH), Christian Resick (LCoB), Andrea Davis (DSPH)
Diagnostics
- Inexpensive, Rapid, Field-Usable Genetic Test to Detect COVID-19 in Asymptomatic Individuals
♦ Wan Shih (Biomed)
Personal Protective Equipment (PPE)
- Biocontainment Intubation “Tent”: Design Refinement, Testing and Deployment
♦ Sharon Walker (CoE), Kara Spiller (Biomed), Caroline Schauer (CoE), Michele Marcolongo (CoE), Amy Throckmorton (Biomed), Michael Waring (CoE), James Lo (CoE)
- Covid-19: Design-Build of “AJflex Shield” For Hospitals and Health Systems in the Philadelphia Region
♦ Amy Throckmorton (Biomed), Michele Marcolongo (CoE), Ellen Bass (CNHP, CCI)
- COVID-19: Developing a PPE Facemask and Validating Its Removal Efficiency
♦ Michael Waring (CoE), Genevieve Dion (WCoMAD)
- PPE Fabric Face Mask for Healthcare Providers and General Use
♦ Genevieve Dion (WCoMAD), Charles Cairns (CoM)
Social and Economic Implications
- Understanding the Disproportionate Impacts of COVID-19 on Low-Income, Minority Communities
♦ Allison Groves (DSPH)
Therapeutics / Vaccines
- Adenosine Deaminase-1 Enhances Efficacy of A DNA Vaccine Encoding SARS-Cov-2 Spike Glycoprotein S1 through Modulation of T Follicular Helper Cells
♦ Michele Kutzler (CoM), Elias Haddad (CoM)
- Irreversible Inactivation of SARS-Cov-2 by Spike Protein Targeting
♦ Irwin Chaiken (CoM)
- SARS-Cov-2 Infection and Sensing in Primary Human Macrophages
♦ Sonia Navas-Martin (CoM), Julio Martin-Garcia (CoM)
A special thanks to our 15 peer reviewers who worked diligently so that we could award these funds quickly.
Though we were unable to fund all projects, we encourage all submitters to seek COVID-19 project funds by searching the subscription funding databases (Funding Institutional and Pivot), signing up for alerts from grants.gov, reaching out federal agencies and foundations, socializing your projects with program managers in those agencies and foundations, and seeking guidance from internal supports through the Drexel Librarians, Institutional Advancement/Foundation Relations, and Office of Research & Innovation. A summary of some opportunities and resources can be found on the ORI's COVID-19 Research Funding Opportunities webpage. Questions? Email researchdevelopment@drexel.edu
- Update on Drexel COVID-19 Monitoring and Restrictions
- Drexel Restricts South Korea for All Travelers
- Italy Added to Restricted Travel List
- And more...
Examples of coronavirus research supported by NIH from NIH RePORTER NIAID support for new research in response to SARS-CoV-2 and COVID-2019 NIAID Funding News, Feb 19, 2020; Notice of Special Interest (NOSI) regarding the Availability of Urgent Competitive Revisions for Research on the 2019 Novel Coronavirus (2019-nCoV) (NOT-AI-20-030).
Please visit the NIH website to learn more.
The platform presenters for Drexel University College of Medicine's annual research day, called Discovery Day, discuss their research topics and tips for success. Read more.
DrExcel Health is proud to present our first podcast episode for the semester with our esteemed guest, Dr. Esther Chernak. Dr. Chernak is the course Director for the Frontiers program at Drexel University College of Medicine, and is the Director of the MD/MPH program. Her work spans over 2 decades of involvement in the Philadelphia health system. The episode can be accessed at anchor.fm.
Six Scientists Share Their Work - Where antiretroviral drugs are taken as prescribed, HIV/AIDS has passed from a crisis to a manageable chronic disease. Researchers are deeply engaged with the next set of questions: How can HIV patients have a better quality of life? What aging conditions or diseases might they be more susceptible to as a result of living with the virus? Are there better treatments that might involve fewer side effects, or that can be used if the existing ones are no longer effective? Can a vaccine be developed that will conquer this health threat once and for all? Read more. Alumni Magazine Spring/Summer 2019

Pictured from left to right: Noreen Robertson, DMD (Associate Vice Dean for Research); Dr. James Allison (2018 Nobel Laureate and 2019 Benjamin Franklin Medal winner); Dr. Brad Jameson (Professor, Department of Biochemistry and Molecular Biology); and Kenny Simansky, PhD (Vice Dean for Research; Professor, Department of Pharmacology and Physiology)
Drexel Students and Faculty Meet Nobel Laureate
Drexel graduate and medical students met with 2018 Nobel Prize winner Dr. James Allison to discuss his work, following his seminar entitled "Immune Checkpoint Blockade in Cancer Therapy: New Insights, Opportunities and Prospects for Cures." Dr. Allison's lecture at Drexel University was held in conjunction with his receipt of the 2019 Benjamin Franklin Medal in Life Sciences. Dr. Allison received the Nobel Prize in Medicine for work on immune checkpoint blockade as a treatment for cancer. He is credited with devising an entirely new approach to cancer therapy and saving many lives.
New Research Solutions Committee for Clinical Research
The Office of the Vice Dean for Research is launching a Research Solutions Committee. The goal is to give clinical researchers an opportunity to address concerns and issues with their clinical research projects.
This will be a monthly meeting, and anyone can request to be placed on the agenda. Then the appropriate parties will be present to discuss and resolve the issue(s). We believe this will bring more transparency into the negotiation process among relevant parties, improve communication, and help us collectively come up with innovative solutions to make our processes more efficient.
The meetings will be held the 2nd Wednesday of every month, 9:30-10:30 a.m., at New College Building, 19th floor, in the Academic Conference Room.
To be added to the agenda, email Janet Matthews BSN, RN, director, Research Program Development, at jm834@drexel.edu. You will be provided with the meeting date and time your subject will be discussed.
Please note that, as always, our office will be available to you for any research concern or issue at any time. The advantage of this committee will be to bring all relevant parties from our office, the Office of Research, and the Office of the Comptroller, as needed, to a common meeting for issues that remain unresolved.
"We're testing the hypothesis that Alzheimer's disease — which perhaps should be called Fischer's disease — is triggered at least in some cases by infection," says Ehrlich, a professor in the Departments of Microbiology & Immunology and Otolaryngology–Head and Neck Surgery. Alumni Magazine (Summer 2018)
Vineet Bhandari, MD, has successfully triggered a process in which cells engulf their own insides in mice subjects, which could be used to prevent chronic lung disease in premature infants. Drexel EXEL Magazine (2018)
A team led by Akhil Vaidya, PhD, has discovered an unusual mechanism that allows two new antimalarial drugs to operate. Drexel EXEL Magazine (2018)
Thousands of U.S. soldiers returned from the 1991 Persian Gulf War with a mysterious, incurable illness. To find answers, College of Medicine researchers are reprogramming veterans' cells. Drexel EXEL Magazine (2018)
A drug compound in development at Drexel would give breast cancer patients the gift of precious time, by keeping metastatic cells from seeding deadly new tumors. Drexel eXel Magazine (2018)
A new study from Drexel researchers sheds light on the parts of the brain that help make a neuron's journey from its birthplace to the brain—and everything that relies on it — possible. Drexel EXEL Magazine (2018)
A battery-powered applicator developed by Michael Weingarten, MD, and Peter Lewin, PhD — as small and light as a watch — is the first portable device to heal chronic wounds with low-frequency ultrasound. Drexel EXEL Magazine (2018)
Joshua Chang Mell, PhD, and colleagues at the University of British Columbia have made progress in understanding how a common pathogen causes the chronic lung infections in cystic fibrosis patients. Drexel EXEL Magazine (2018)
Thomas Trojian, MD, and colleagues believe youth coaches should teach young athletes better movement techniques that will reduce lower-body injuries. Drexel EXEL Magazine (2018)
Thousands of U.S. soldiers returned from the 1991 Persian Gulf War with a mysterious, incurable illness. To find answers, College of Medicine researchers are reprogramming veterans' cells. Drexel eXel Magazine (2018)
Researchers from Drexel University College of Medicine and the University of Texas at Austin improved respiratory function in rodents with spinal cord injuries after successfully transplanting a special class of neural cells, called V2a interneurons. Their results, published this week in the Journal of Neurotrauma, indicate that these lab-grown cells have the potential to one day help paralyzed patients breathe without a ventilator. Science Magazine / Drexel News
Molecular & Cell Biology & Genetics PhD Maya Rao recently published "Interaction between the AAA+ ATPase p97 and its cofactor ataxin3 in health and disease: Nucleotide-induced conformational changes regulate cofactor binding" in the November issue of the Journal of Biological Chemistry. (February 2018)
A $25,000 grant for an HIV Cancer Pilot Award from the Sidney Kimmel Cancer Center will support a collaborative investigation into the severity of anal dysplasia, which can lead to anal cancer, and its association with inflammation in HIV infection. (November 2017)
With topics ranging from HIV/AIDS to spinal cord injury and neuroengineering, more than 380 Drexel University College of Medicine students and scientists showcased original research at Discovery Day 2017 on Thursday, Oct. 12.
STAT — a Boston-based national publication focused on science and health news — has named Halley Oyer, PhD, one of the "brightest young minds in life science." Oyer is a College of Medicine postdoc working in the laboratory of Felix Kim, PhD. (October 2017)
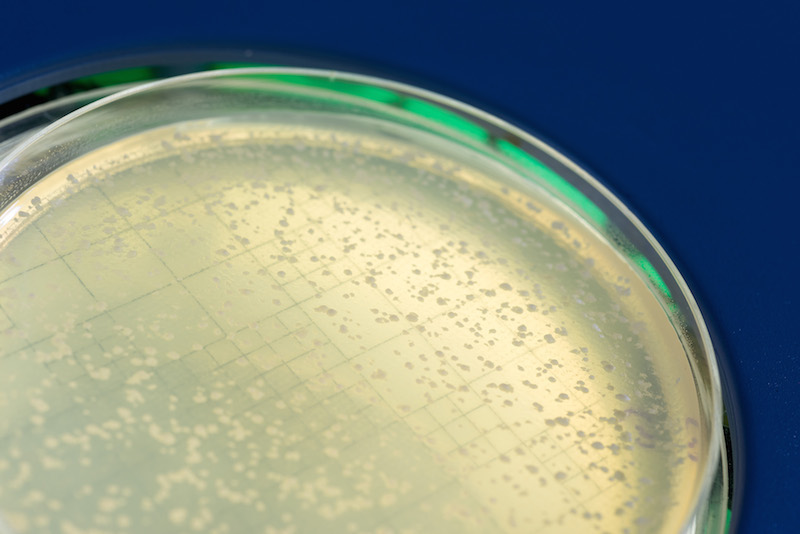
A College of Medicine study reveals an unexpected function of the homologous recombination protein Rad52 and may help to identify new therapeutic targets for cancer. (June 8, 2017)

College of Medicine researchers shed light on the neurological consequences of exposure to low-levels of nerve agents and suggest that drugs like tubacin could treat some of the toxins’ neurological effects. (June 6, 2017)

Researchers at Howard Hughes Medical Institute and the Eunice Kennedy Shriver National Institute for Child Health and Human Development are getting a first glimpse at the inner-workings of live cells thanks to a new microscopy technique pioneered by Nobel laureate Eric Betzig with help from engineers at Drexel University. Their method uses grids of light that activate fluorescent color tags on each type of organelle — the result is a 3-D video that gives researchers their best look at how cells function. It will allow scientists to better understand how cells react to environmental stressors and respond to drug treatment. (May 24, 2017)

Three biopharmaceutical startups led by Drexel University researchers are one step closer to bringing new, potentially life-saving drugs to the market. (May 10, 2017)
Scientists have made new headway in understanding how a deadly pathogen evolves during chronic lung infections in cystic fibrosis patients. "By looking at changes in the genome over time, we were able to see patterns — common themes that help us to better understand how this particular species evolves in its environment and how CF patients become chronically infected," said study co-corresponding author Joshua Chang Mell, PhD, an assistant professor at Drexel University College of Medicine. Drexel Now (March 21, 2017)
Many thanks to the faculty and staff who participated in the 2017 Commonwealth Universal Research Enhancement (CURE) grant program competition. After an internal call for proposals, a scientific review panel met on March 3, 2017, to evaluate the applications that were submitted. The panel comprised faculty members from across the University. (March 20, 2017)
For researchers at Drexel University College of Medicine, the acquisition of two new confocal microscopes provides an unprecedented view into the human body — and fresh insights into conditions such as autism, neuroinflammation, HIV-associated neurocognitive impairment, heart attacks, cancer and more. Pulse (Spring 2017)
Back to Top